From RNA-seq to chemical enrichment: a complete workflow
Luigi Corsaro
2026-07-07
Source:vignettes/articles/tutorial_rnaseq_workflow.Rmd
tutorial_rnaseq_workflow.RmdBiological context
Dexamethasone is a synthetic glucocorticoid widely used as an anti-inflammatory and immunosuppressive agent. Its transcriptomic effects on immune cells are well characterised: it suppresses pro-inflammatory cytokines, reprograms monocyte and lymphocyte gene programmes, and leaves a distinctive signature in peripheral blood mononuclear cells (PBMCs).
This tutorial asks a straightforward question: given the RNA-seq signature of dexamethasone treatment in human PBMCs, which chemicals in the Comparative Toxicogenomics Database (CTD) share that signature?
We answer it using all four methods provided by ctdR — GSEA, ORA, CAMERA, and GSVA — and show how to visualise and interpret each result.
Data source: we use GSE311566, a public GEO series comparing the effects of PFAS chemicals and dexamethasone on human PBMCs from female donors. We work with the Dexamethasone vs. vehicle (DMSO/Ctrl) contrast, female samples only (7 samples total: 4 Ctrl + 3 Dex).
What this tutorial covers
- Downloading and preparing public RNA-seq count data from GEO
- Differential expression with
limma- Chemical enrichment analysis (GSEA, ORA, CAMERA, GSVA) using
ctdR- Visualisation with
plot_CTD()What this tutorial does NOT cover
- Raw read quality control (FastQC, trimming)
- Alignment and count generation
- Normalisation strategy selection (TMM, VST, etc.)
- Batch correction or covariate adjustment
- Biological replicate adequacy or power analysis
This is an end-to-end illustration of the
ctdRworkflow on publicly available processed data, not a template for a production differential expression pipeline.
Step 1 — Download data from GEO
The normalised count matrix for GSE311566 is available directly from NCBI FTP. We download it once and cache it in the session temporary directory; re-running this chunk skips the download if the file already exists.
GEO_URL <- paste0(
"https://ftp.ncbi.nlm.nih.gov/geo/series/GSE311nnn/GSE311566/",
"suppl/GSE311566_PBMCs_Female_normalized_counts.txt.gz"
)
tmp_gz <- file.path(tempdir(), "GSE311566_Female_normalized_counts.txt.gz")
if (!file.exists(tmp_gz)) {
message("Downloading GSE311566 from NCBI FTP ...")
utils::download.file(GEO_URL, tmp_gz, mode = "wb", quiet = FALSE)
}Step 2 — Prepare the expression matrix
counts <- utils::read.table(
gzfile(tmp_gz),
header = TRUE, sep = "\t", row.names = 1,
check.names = FALSE, stringsAsFactors = FALSE
)
counts$DESCRIPTION <- NULL # all NA in this dataset
# Female Ctrl and DEX samples only
keep_cols <- grep("^(Ctrl|DEX)_F_", colnames(counts), value = TRUE)
counts <- counts[, keep_cols]
group <- factor(
ifelse(grepl("^DEX_", keep_cols), "Dex", "DMSO"),
levels = c("DMSO", "Dex")
)
message(nrow(counts), " genes, ", ncol(counts), " samples: ",
paste(table(group), names(table(group)), sep = " ", collapse = " / "))The GEO file uses Ensembl gene IDs. We strip version suffixes and map
to Entrez IDs, which are the identifier type required by
ctdR.
ensg <- sub("\\..*$", "", rownames(counts))
entrez_map <- AnnotationDbi::mapIds(
org.Hs.eg.db,
keys = ensg,
keytype = "ENSEMBL",
column = "ENTREZID",
multiVals = "first"
)
keep_g <- !is.na(entrez_map) & !duplicated(entrez_map)
counts <- counts[keep_g, ]
rownames(counts) <- entrez_map[keep_g]
expr <- log2(as.matrix(counts) + 1)
message(nrow(expr), " genes retained after Entrez mapping")Step 3 — Import CTD data
ctd_csv <- "~/Downloads/CTD_chem_gene_ixns.csv"
ctd_gz <- "~/Downloads/CTD_chem_gene_ixns.csv.gz"
if (file.exists(ctd_csv)) {
message("Using full CTD: ", ctd_csv)
import_CTD(ctd_csv)
} else if (file.exists(ctd_gz)) {
message("Using full CTD (compressed): ", ctd_gz)
import_CTD(ctd_gz)
} else {
message(
"Full CTD not found in ~/Downloads/ — using bundled toy sample ",
"(10 chemicals). Results will be illustrative only.\n",
"Download CTD_chem_gene_ixns.csv.gz from ctdbase.org for a ",
"powered analysis."
)
import_CTD(system.file(
"extdata", "CTD_chem_gene_ixns_sample.csv",
package = "ctdR"
))
}Note on CTD coverage: this tutorial uses the small toy dataset bundled with
ctdR(10 chemicals, 17 genes). It is sufficient to demonstrate the API and visualisations. For a powered analysis capable of discovering novel chemical associations, download the fullCTD_chem_gene_ixns.csv.gzfrom https://ctdbase.org/reports/CTD_chem_gene_ixns.csv.gz and runimport_CTD()on that file instead.
Step 4 — Differential expression with limma
We fit a linear model and apply empirical Bayes moderation. The
design matrix has two columns: the intercept (DMSO baseline) and
groupDex (Dex vs DMSO contrast, coefficient 2).
design <- model.matrix(~ group)
colnames(design)
#> [1] "(Intercept)" "groupDex"
fit <- limma::lmFit(expr, design)
fit <- limma::eBayes(fit)
de <- limma::topTable(fit, coef = 2, n = Inf, sort.by = "P")
de$EntrezID <- rownames(de)
head(de[, c("EntrezID", "logFC", "t", "P.Value", "adj.P.Val")])
#> EntrezID logFC t P.Value adj.P.Val
#> 2289 2289 2.8860368 66.81282 5.381612e-09 0.000200056
#> 64744 64744 1.5371202 33.39116 2.148090e-07 0.002665942
#> 8553 8553 -1.2969205 -33.38130 2.151457e-07 0.002665942
#> 8761 8761 0.7060472 27.55983 5.941309e-07 0.005521555
#> 8395 8395 -0.9214413 -25.57857 8.817929e-07 0.006555954
#> 78999 78999 0.9863471 23.27464 1.452352e-06 0.008998287A brief look at significance:
Step 5 — Chemical enrichment
GSEA — Gene Set Enrichment Analysis
GSEA uses the full ranked gene list to detect
chemicals whose known target genes collectively cluster toward one
extreme of the ranking, without requiring a significance threshold. We
supply the moderated t-statistic from limma as the stat
column so that directionality (up vs down) is preserved and ties at
non-significant genes are avoided. Pass nproc = N to
parallelise across N cores.
gsea_input <- data.frame(
EntrezID = de$EntrezID,
pvalue = de$P.Value,
stat = de$t, # signed ranking: positive = up in Dex
row.names = NULL
)
gsea_results <- enrichment_CTD(
gsea_input, method = "GSEA",
minSize = 3, maxSize = 500 # gene set size filter
)
head(gsea_results[, c(
"ChemicalName", "PValue", "PValueAdjusted",
"NormalizedEnrichmentScore", "FoldEnrichment"
)])
#> ChemicalName PValue PValueAdjusted NormalizedEnrichmentScore
#> 1 Estradiol 0.02328381 0.2328381 -1.4470219
#> 2 Acetaminophen 0.74846626 0.9355828 0.7939608
#> 3 Benzo(a)pyrene 0.71342685 0.9355828 0.8077455
#> 4 Cadmium 0.73456790 0.9355828 0.7949407
#> 5 Cisplatin 0.73630832 0.9355828 0.7844192
#> 6 Cyclophosphamide 0.73809524 0.9355828 -0.8136112
#> FoldEnrichment
#> 1 33.33723
#> 2 15.17452
#> 3 16.02867
#> 4 16.87423
#> 5 15.08997
#> 6 19.82472FoldEnrichment here is |ES| / mean(|ES|)
across all tested chemicals — a relative measure of how strongly
enriched this chemical’s gene set is compared to the average.
NES (Normalized Enrichment Score) accounts for gene set
size differences and is the primary statistic for ranking chemicals.
plot_CTD(gsea_results, type = "bar", n = 10,
title = "GSEA — top chemicals by NES")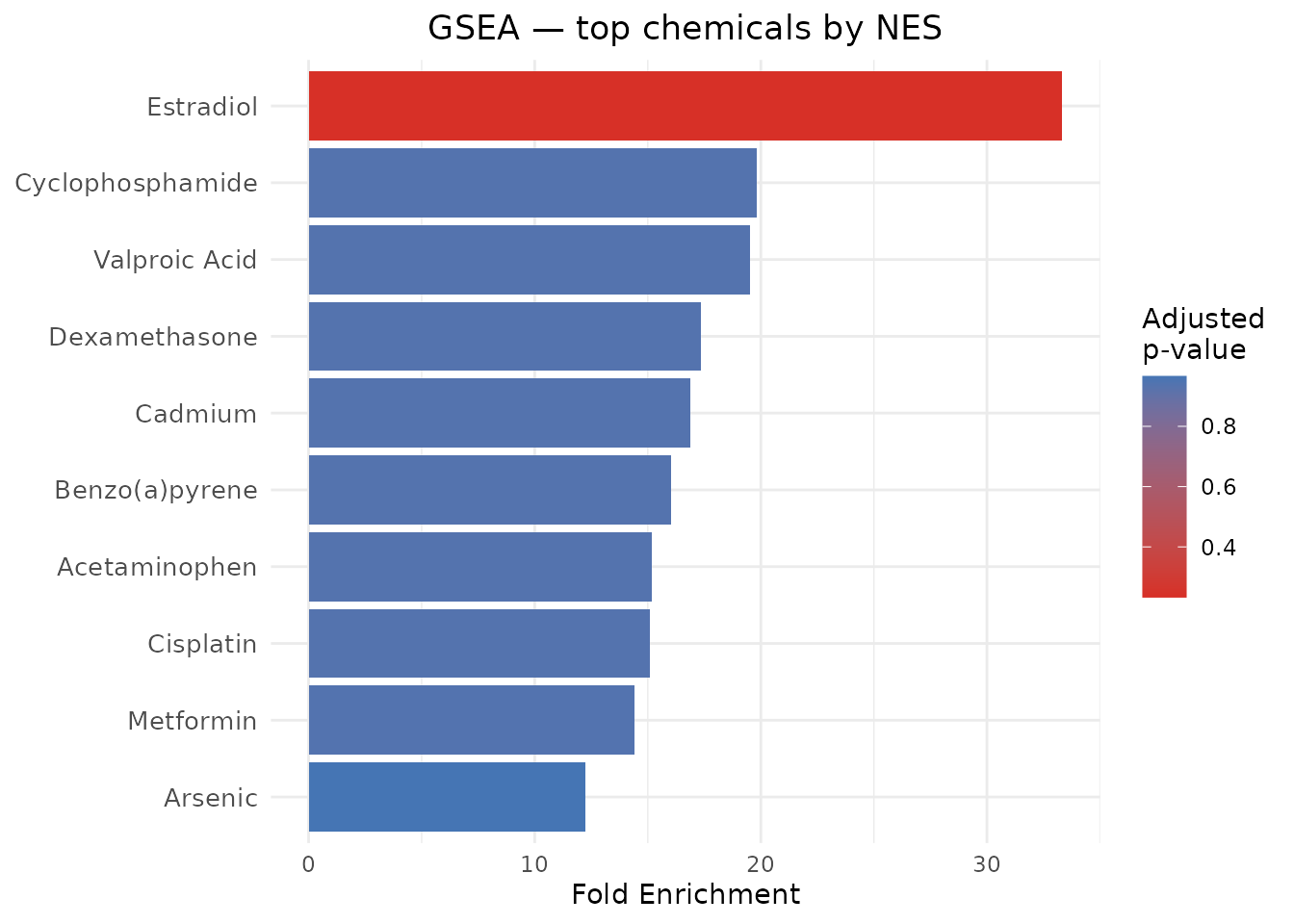
plot_CTD(gsea_results, type = "dot", n = 10,
title = "GSEA — top chemicals (dot)")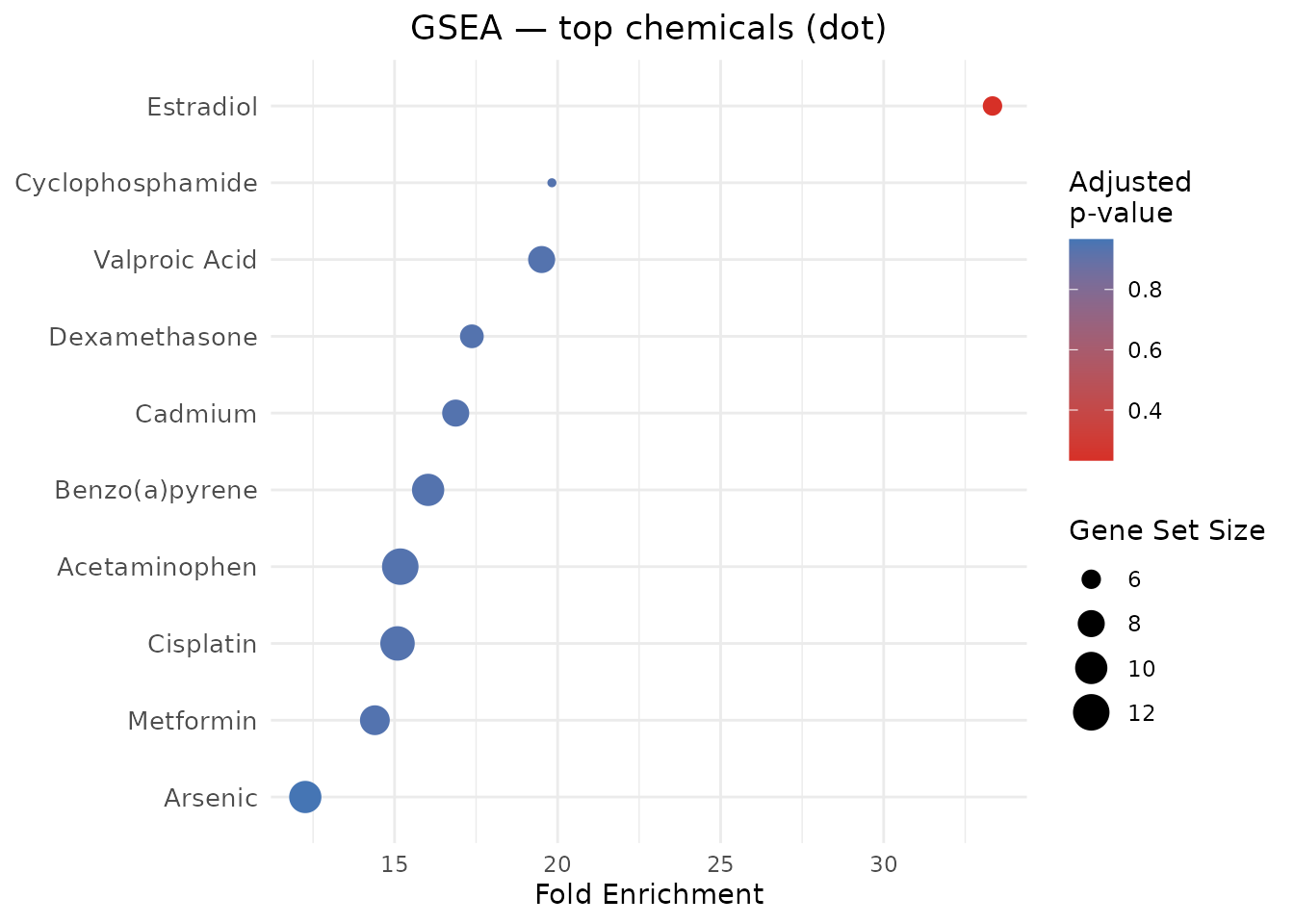
Where is Dexamethasone?
The top-ranked chemical may or may not be Dexamethasone depending on which CTD dataset is loaded. With the full CTD (~11 000 chemicals and strict BH correction) Dexamethasone competes against many other chemicals and may rank lower than expected, or may not reach significance after correction. The code below locates it regardless of its rank:
dex_idx <- grep("dexamethasone", gsea_results$ChemicalName,
ignore.case = TRUE)
if (length(dex_idx) > 0) {
cat("Dexamethasone: rank", dex_idx, "of", nrow(gsea_results), "\n")
gsea_results[dex_idx, c("ChemicalName", "PValue", "PValueAdjusted",
"NormalizedEnrichmentScore", "FoldEnrichment")]
} else {
message("Dexamethasone not found — it may be absent from the loaded CTD cache.")
}
#> Dexamethasone: rank 7 of 10
#> ChemicalName PValue PValueAdjusted NormalizedEnrichmentScore
#> 7 Dexamethasone 0.7396694 0.9355828 0.7902494
#> FoldEnrichment
#> 7 17.37013Why Dexamethasone may not top the list with the full CTD:
- Multiple testing burden — 11 000 tests vs 10 in the toy sample; the BH threshold is ~1 000× more stringent.
- Larger gene sets — in the full CTD, Dexamethasone has hundreds of annotated targets; very large sets dilute the GSEA signal in small cohorts (7 samples here).
- Competing chemicals — other chemicals may have gene sets that overlap more tightly with the top-ranked DE genes.
Direction-aware GSEA with interaction_types
The standard GSEA above uses all CTD interactions regardless of
direction. With interaction_types we can restrict each
chemical’s gene set to interactions of a specific type, creating a
concordance test: do chemicals that are known to
increase gene expression share targets with genes we observe
up-regulated?
# Chemicals whose INCREASE-expression targets are enriched among up-regulated genes
gsea_up <- enrichment_CTD(
gsea_input, method = "GSEA",
minSize = 3, maxSize = 500,
interaction_types = "increases^expression"
)
# Chemicals whose DECREASE-expression targets are enriched among down-regulated genes
# Flip stat sign so down-regulated genes (negative t) rank at the top
gsea_input_down <- transform(gsea_input, stat = -stat)
gsea_down <- enrichment_CTD(
gsea_input_down, method = "GSEA",
minSize = 3, maxSize = 500,
interaction_types = "decreases^expression"
)
# Chemicals consistent with Dex treatment (up or down, direction-matched)
message("Direction-aware GSEA — increases^expression: ",
nrow(gsea_up), " chemicals tested")
message("Direction-aware GSEA — decreases^expression: ",
nrow(gsea_down), " chemicals tested")A chemical appearing in both gsea_up
and gsea_down with positive NES in each is particularly
strong evidence of a concordant transcriptomic footprint.
ORA — Over-Representation Analysis
ORA works on a binary gene list: genes declared significant vs the rest of the universe. It asks whether the overlap between your significant genes and each chemical’s known targets is larger than expected by chance (hypergeometric test).
sig <- de[de$adj.P.Val < 0.05, c("EntrezID", "P.Value")]
colnames(sig)[2] <- "pvalue"
message(nrow(sig), " genes at adj.P.Val < 0.05")
ora_results <- enrichment_CTD(
sig, method = "ORA",
universe = de$EntrezID, # restrict background to measured genes
minGSSize = 3,
maxGSSize = 500
)
if (nrow(ora_results) > 0) {
head(ora_results[, c(
"ChemicalName", "PValue", "PValueAdjusted",
"FoldEnrichment", "Count", "EnrichedGenes"
)])
} else {
message(
"ORA returned 0 results — expected with the toy CTD sample.\n",
"Run import_CTD() on the full CTD file for a powered analysis."
)
}
if (nrow(ora_results) > 0) {
dex_idx <- grep("dexamethasone", ora_results$ChemicalName,
ignore.case = TRUE)
if (length(dex_idx) > 0) {
cat("Dexamethasone: rank", dex_idx, "of", nrow(ora_results), "\n")
ora_results[dex_idx, c("ChemicalName", "PValue", "PValueAdjusted",
"FoldEnrichment", "Count")]
} else {
message("Dexamethasone not found in ORA results.")
}
}
plot_CTD(ora_results, type = "dot", n = 10,
title = "ORA — top chemicals by fold enrichment")FoldEnrichment in ORA is
GeneRatio / BackgroundRatio: the proportion of your
significant genes that are targets of this chemical, divided by the same
proportion in the background universe. A value greater than 1 indicates
over-representation.
CAMERA — Competitive gene-set test
CAMERA tests, for each chemical, whether its target genes show a stronger differential signal than the rest of the transcriptome, while correcting for inter-gene correlation within the gene set. It is particularly well suited to expression matrix experiments with a clear design and contrast.
CAMERA does not consume a pre-computed DEG result: it takes the
full expression matrix plus a
design and a
contrast, and recomputes the differential
signal internally. The chemical→gene “index” is built by
ctdR from CTD targets. We reuse the design
matrix built for limma; here design carries the whole
layout while contrast = 2 selects which
coefficient to test — the groupDex effect (Dex vs
DMSO).
camera_results <- enrichment_CTD(
expr,
method = "CAMERA",
design = design,
contrast = 2
)
head(camera_results[, c(
"ChemicalName", "PValue", "PValueAdjusted",
"GeneSetSize", "Direction"
)])
#> ChemicalName PValue PValueAdjusted GeneSetSize Direction
#> 1 Estradiol 0.02495925 0.2495925 6 Down
#> 2 Acetaminophen 0.26645058 0.5007785 12 Up
#> 3 Benzo(a)pyrene 0.31813538 0.5007785 10 Up
#> 4 Cadmium 0.16732813 0.5007785 8 Up
#> 5 Cisplatin 0.31687483 0.5007785 11 Up
#> 6 Dexamethasone 0.33074736 0.5007785 7 UpUnlike ORA and GSEA, CAMERA does not report a
FoldEnrichment value — it reports Direction
("Up" or "Down"), indicating whether the
chemical’s target genes are predominantly up- or down-regulated under
the tested contrast.
plot_CTD(camera_results, type = "bar", n = 10,
title = "CAMERA — direction-aware chemical enrichment")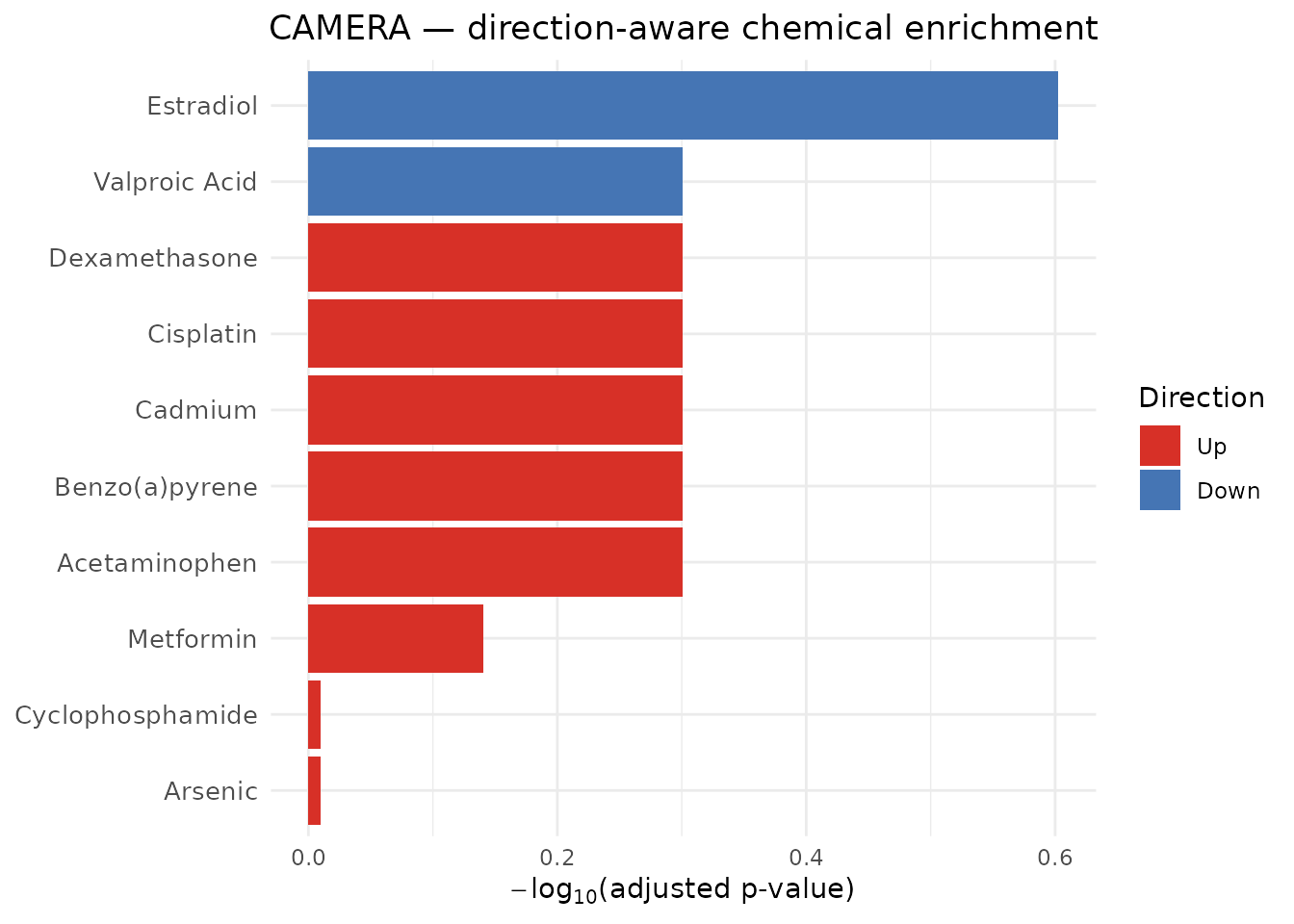
dex_idx <- grep("dexamethasone", camera_results$ChemicalName,
ignore.case = TRUE)
if (length(dex_idx) > 0) {
cat("Dexamethasone: rank", dex_idx, "of", nrow(camera_results), "\n")
camera_results[dex_idx, c("ChemicalName", "PValue", "PValueAdjusted",
"GeneSetSize", "Direction")]
} else {
message("Dexamethasone not found in CAMERA results.")
}
#> Dexamethasone: rank 6 of 10
#> ChemicalName PValue PValueAdjusted GeneSetSize Direction
#> 6 Dexamethasone 0.3307474 0.5007785 7 UpGSVA — Gene Set Variation Analysis
GSVA produces a per-sample enrichment score for each chemical: a matrix with chemicals in rows and samples in columns. There is no single group-level p-value; instead, these scores can be used for clustering, heatmap visualisation, survival association, or as input to downstream tests.
Computation time: GSVA scores all chemicals across all samples. On the full expression matrix this may take a few minutes. Use
BiocParallel::register()to speed it up on multi-core machines.
gsva_scores <- enrichment_CTD(
expr, method = "GSVA",
minSize = 3, maxSize = 500
)
dim(gsva_scores) # chemicals x samples
#> [1] 10 7
plot_CTD(gsva_scores,
title = "GSVA — per-sample chemical enrichment scores")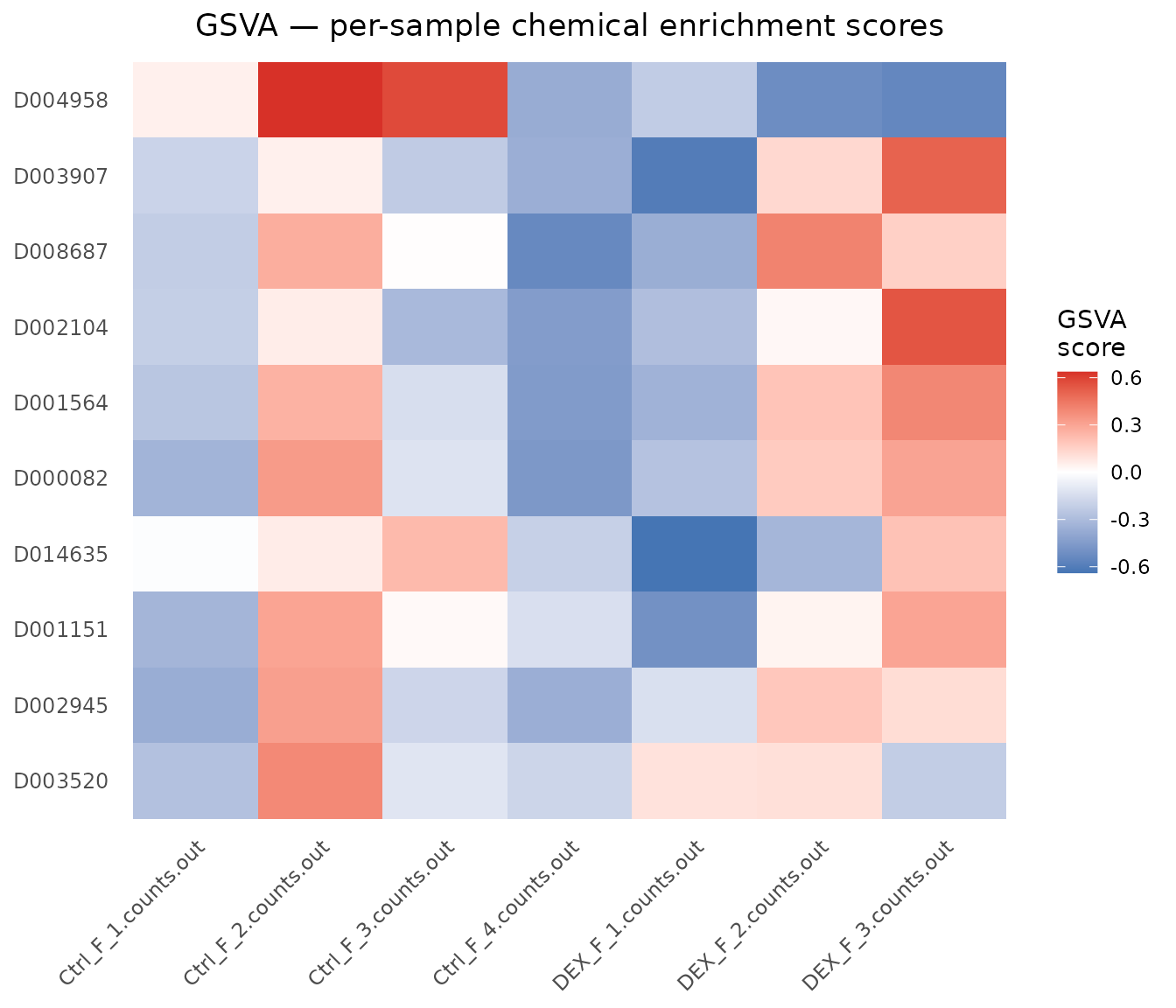
The heatmap rows are chemicals ranked by across-sample variance (top-variance chemicals shown first). Samples that cluster together share a similar chemical-exposure transcriptomic profile.
To inspect Dexamethasone’s per-sample scores directly:
dex_idx <- grep("dexamethasone", rownames(gsva_scores),
ignore.case = TRUE)
if (length(dex_idx) > 0) {
cat("Dexamethasone row(s):", rownames(gsva_scores)[dex_idx], "\n")
gsva_scores[dex_idx, , drop = FALSE]
} else {
message("Dexamethasone not found in GSVA score matrix.")
}Positive scores indicate that Dexamethasone’s target genes are collectively up-regulated in that sample relative to the background; negative scores indicate down-regulation. A clear separation between Dex and Ctrl samples (positive scores in Dex, negative in Ctrl, or vice versa) would support the expected glucocorticoid signature.
Step 6 — Comparing results across methods
Each method answers a subtly different question:
| Method | Input | Question answered | Key output column |
|---|---|---|---|
| GSEA | Full ranked list | Do this chemical’s targets cluster at the extreme of the ranking? |
NES, FoldEnrichment
|
| ORA | Significant gene list | Is the overlap larger than expected by chance? |
FoldEnrichment, Count
|
| CAMERA | Expression matrix + design | Is the differential signal stronger than the transcriptome average? | Direction |
| GSVA | Expression matrix | What is the per-sample enrichment score? | score matrix |
GSEA and ORA both report FoldEnrichment, but it means
different things: in GSEA it is relative to the mean ES across chemicals
(|ES| / mean(|ES|)); in ORA it is the ratio of observed to
expected overlap (GeneRatio / BackgroundRatio). Do not
compare these numbers across methods.
For a discovery workflow on a single contrast, GSEA is recommended as the primary method — it uses all genes and is robust to the choice of significance threshold. ORA is useful as a complementary check when a reliable binary gene list is available. CAMERA is the method of choice when inter-gene correlation is a concern. GSVA is best reserved for multi-sample stratification or when you want to visualise chemical signatures across a cohort.
Dexamethasone — ranking recap across all methods
Since Dexamethasone is the treatment used in GSE311566, we expect its CTD gene set to carry a strong signal in this dataset. The code below collects its position and key statistics from each method into a single summary, regardless of whether it reached the top rank.
dex_gsea <- local({
idx <- which(tolower(gsea_results$ChemicalName) == "dexamethasone")
if (length(idx) == 0L) return(NULL)
r <- gsea_results[idx, ]
data.frame(
Method = "GSEA",
Rank = idx,
Total = nrow(gsea_results),
PValue = signif(r$PValue, 3),
PAdj = signif(r$PValueAdjusted, 3),
StatLabel = "NES",
Stat = signif(r$NormalizedEnrichmentScore, 3)
)
})
dex_ora <- local({
if (nrow(ora_results) == 0L) return(NULL)
idx <- which(tolower(ora_results$ChemicalName) == "dexamethasone")
if (length(idx) == 0L) return(NULL)
r <- ora_results[idx, ]
data.frame(
Method = "ORA",
Rank = idx,
Total = nrow(ora_results),
PValue = signif(r$PValue, 3),
PAdj = signif(r$PValueAdjusted, 3),
StatLabel = "FoldEnrichment",
Stat = signif(r$FoldEnrichment, 3)
)
})
dex_camera <- local({
idx <- which(tolower(camera_results$ChemicalName) == "dexamethasone")
if (length(idx) == 0L) return(NULL)
r <- camera_results[idx, ]
data.frame(
Method = "CAMERA",
Rank = idx,
Total = nrow(camera_results),
PValue = signif(r$PValue, 3),
PAdj = signif(r$PValueAdjusted, 3),
StatLabel = "Direction",
Stat = r$Direction
)
})
dex_gsva <- local({
# GSVA rownames are ChemicalIDs (e.g. "D003907"), not names.
# Load the chemicals metadata to map name -> ID.
e <- new.env(parent = emptyenv())
load(file.path(rappdirs::user_cache_dir("ctdR"), "chemicals.rda"),
envir = e)
dex_id <- e$chemicals$ChemicalID[
tolower(e$chemicals$ChemicalName) == "dexamethasone"
]
if (length(dex_id) == 0L || !dex_id %in% rownames(gsva_scores))
return(NULL)
scores <- gsva_scores[dex_id, , drop = FALSE]
dex_samples <- grepl("^DEX_", colnames(gsva_scores))
ctrl_samples <- grepl("^Ctrl_", colnames(gsva_scores))
mean_dex <- mean(scores[, dex_samples])
mean_ctrl <- mean(scores[, ctrl_samples])
data.frame(
Method = "GSVA",
Rank = NA_integer_,
Total = nrow(gsva_scores),
PValue = NA_real_,
PAdj = NA_real_,
StatLabel = "mean(Dex) - mean(Ctrl)",
Stat = signif(mean_dex - mean_ctrl, 3)
)
})
recap <- do.call(rbind, Filter(Negate(is.null),
list(dex_gsea, dex_ora, dex_camera, dex_gsva)))
if (is.null(recap) || nrow(recap) == 0L) {
message("Dexamethasone not found in any method's results.")
} else {
recap$RankOf <- ifelse(
is.na(recap$Rank), "—",
paste0(recap$Rank, " / ", recap$Total)
)
print(recap[, c("Method", "RankOf", "PValue", "PAdj",
"StatLabel", "Stat")],
row.names = FALSE)
}
#> Method RankOf PValue PAdj StatLabel Stat
#> GSEA 7 / 10 0.740 0.936 NES 0.79
#> CAMERA 6 / 10 0.331 0.501 Direction Up
#> GSVA — NA NA mean(Dex) - mean(Ctrl) 0.195RankOf is the position of Dexamethasone in each result
table sorted by adjusted p-value (GSEA, ORA, CAMERA) or — for GSVA — the
difference between the mean per-sample score in treated vs control
samples (positive = target genes up-regulated in Dex samples).
Session information
sessionInfo()
#> R version 4.6.1 (2026-06-24)
#> Platform: x86_64-pc-linux-gnu
#> Running under: Ubuntu 24.04.4 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
#>
#> locale:
#> [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
#> [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
#> [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
#> [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
#>
#> time zone: UTC
#> tzcode source: system (glibc)
#>
#> attached base packages:
#> [1] stats4 stats graphics grDevices utils datasets methods
#> [8] base
#>
#> other attached packages:
#> [1] org.Hs.eg.db_3.23.1 AnnotationDbi_1.74.0 IRanges_2.46.0
#> [4] S4Vectors_0.50.1 Biobase_2.72.0 BiocGenerics_0.58.1
#> [7] generics_0.1.4 limma_3.68.4 ctdR_0.99.6
#> [10] BiocStyle_2.40.0
#>
#> loaded via a namespace (and not attached):
#> [1] splines_4.6.1 ggplotify_0.1.3
#> [3] tibble_3.3.1 polyclip_1.10-7
#> [5] enrichit_0.2.0 graph_1.90.0
#> [7] XML_3.99-0.23 lifecycle_1.0.5
#> [9] httr2_1.2.3 processx_3.9.0
#> [11] lattice_0.22-9 vroom_1.7.1
#> [13] MASS_7.3-65 magrittr_2.0.5
#> [15] sass_0.4.10 rmarkdown_2.31
#> [17] jquerylib_0.1.4 yaml_2.3.12
#> [19] otel_0.2.0 ggtangle_0.1.2
#> [21] cowplot_1.2.0 DBI_1.3.0
#> [23] RColorBrewer_1.1-3 abind_1.4-8
#> [25] GenomicRanges_1.64.0 purrr_1.2.2
#> [27] yulab.utils_0.2.4 tweenr_2.0.3
#> [29] rappdirs_0.3.4 aisdk_1.4.12
#> [31] gdtools_0.5.1 enrichplot_1.32.0
#> [33] ggrepel_0.9.8 irlba_2.3.7
#> [35] tidytree_0.4.8 GSVA_2.6.2
#> [37] annotate_1.90.0 pkgdown_2.2.0
#> [39] DelayedMatrixStats_1.34.0 codetools_0.2-20
#> [41] DelayedArray_0.38.2 DOSE_4.6.0
#> [43] ggforce_0.5.0 tidyselect_1.2.1
#> [45] aplot_0.3.0 memuse_4.2-3
#> [47] farver_2.1.2 ScaledMatrix_1.20.0
#> [49] matrixStats_1.5.0 Seqinfo_1.2.0
#> [51] jsonlite_2.0.0 systemfonts_1.3.2
#> [53] tools_4.6.1 ggnewscale_0.5.2
#> [55] treeio_1.36.1 ragg_1.5.2
#> [57] Rcpp_1.1.2 glue_1.8.1
#> [59] SparseArray_1.12.2 xfun_0.59
#> [61] qvalue_2.44.0 MatrixGenerics_1.24.0
#> [63] dplyr_1.2.1 HDF5Array_1.40.0
#> [65] withr_3.0.3 BiocManager_1.30.27
#> [67] fastmap_1.2.0 rhdf5filters_1.24.0
#> [69] callr_3.8.0 digest_0.6.39
#> [71] rsvd_1.0.5 R6_2.6.1
#> [73] gridGraphics_0.5-1 textshaping_1.0.5
#> [75] GO.db_3.23.1 RSQLite_3.53.3
#> [77] h5mread_1.4.0 tidyr_1.3.2
#> [79] fontLiberation_0.1.0 data.table_1.18.4
#> [81] httr_1.4.8 htmlwidgets_1.6.4
#> [83] S4Arrays_1.12.0 scatterpie_0.2.6
#> [85] pkgconfig_2.0.3 gtable_0.3.6
#> [87] blob_1.3.0 S7_0.2.2
#> [89] SingleCellExperiment_1.34.0 XVector_0.52.0
#> [91] clusterProfiler_4.20.0 htmltools_0.5.9
#> [93] fontBitstreamVera_0.1.1 bookdown_0.47
#> [95] fgsea_1.38.0 GSEABase_1.74.0
#> [97] scales_1.4.0 png_0.1-9
#> [99] SpatialExperiment_1.22.0 ggfun_0.2.1
#> [101] knitr_1.51 rjson_0.2.23
#> [103] tzdb_0.5.0 reshape2_1.4.5
#> [105] nlme_3.1-169 cachem_1.1.0
#> [107] rhdf5_2.56.0 stringr_1.6.0
#> [109] parallel_4.6.1 desc_1.4.3
#> [111] pillar_1.11.1 grid_4.6.1
#> [113] vctrs_0.7.3 BiocSingular_1.28.0
#> [115] tidydr_0.0.6 beachmat_2.28.0
#> [117] xtable_1.8-8 cluster_2.1.8.2
#> [119] evaluate_1.0.5 magick_2.9.1
#> [121] readr_2.2.0 cli_3.6.6
#> [123] compiler_4.6.1 rlang_1.3.0
#> [125] crayon_1.5.3 labeling_0.4.3
#> [127] ps_1.9.3 plyr_1.8.9
#> [129] fs_2.1.0 ggiraph_0.9.6
#> [131] stringi_1.8.7 BiocParallel_1.46.0
#> [133] Biostrings_2.80.1 lazyeval_0.2.3
#> [135] GOSemSim_2.38.3 fontquiver_0.2.1
#> [137] Matrix_1.7-5 hms_1.1.4
#> [139] patchwork_1.3.2 sparseMatrixStats_1.24.0
#> [141] bit64_4.8.2 ggplot2_4.0.3
#> [143] Rhdf5lib_2.0.0 KEGGREST_1.52.2
#> [145] statmod_1.5.2 SummarizedExperiment_1.42.0
#> [147] igraph_2.3.3 memoise_2.0.1
#> [149] bslib_0.11.0 ggtree_4.2.0
#> [151] fastmatch_1.1-8 bit_4.6.0
#> [153] ape_5.8-1 gson_0.2.0